
Tidal volume in acute respiratory distress syndrome: how best to select it
Introduction
The wide majority of critically ill patients are subject to invasive mechanical ventilation during their stay in the intensive care unit, and patients with acute respiratory distress syndrome (ARDS) are almost invariably managed by invasive mechanical ventilation. Despite extensive research over nearly half a century, no specific therapy exists for ARDS, and mechanical ventilation remains the key form of supportive care (1). In fact, mechanical ventilation per se is not a cure for ARDS; it works by simply buying time by maintaining a sufficient gas exchange for patient survival. This effect is achieved by taking over the function of patients’ respiratory muscles (2). In ARDS, minute ventilation is increased to a level that is much greater than in healthy subjects, due to an abnormally increased respiratory drive (3) and the elevated amount of pulmonary dead space (4). Indeed, the respiratory muscles of patients with ARDS are unable, for several reasons, to drive lung ventilation to a level which is enough to meet patients’ need. Then, the effect on oxygenation provided by mechanical ventilation is dual: it allows for an accurate titration of the fraction of oxygen in the inspired gas, and it provides an inspiratory pressure which is enough to open some of the collapsed lung units, thus making it possible for the blood passing through these regions to be oxygenated during the inspiratory phase. On the other hand, it is the ventilation delivered by the mechanical ventilation, which is allows for carbon dioxide (CO2) elimination.
Unfortunately, a completely “safe” lung ventilation does not exist, and the main side-effects related to mechanical ventilation are the hemodynamic instability secondary to the increased intrathoracic pressures and the mechanical trauma to the lung structure. Indeed, mechanical ventilation itself can bring to a further damage of the lung, through the activation of an inflammatory response and it has been demonstrated that, even in the absence of a pre-existing lung injury, mechanical ventilation may lead to the development of ventilator-induced lung injury (VILI). As a consequence, the so-called “protective ventilation” is an approach which aims to an individual tailoring of ventilatory support, according to the best compromise among respiratory mechanics, recruitability, gas exchange and hemodynamics. The present review deals with the physiological effects of delivering a certain tidal volume to the lungs of patients with ARDS, and suggests an approach to tidal volume selection.
Tidal volume and VILI
Experimental evidence has accumulated in the last 30 years showing that mechanical ventilation provided with elevated volumes and/or pressures can directly injure the lung (5). The current knowledge suggests that ventilation with elevated tidal volumes lead to a process of alveolar disruption that in turn may trigger the chain of events described below.
The consequences of high tidal volume ventilation were termed, once recognized, “volotrauma” (6) and this has been the ground for subsequent clinical trials that established the low tidal volume approach to mechanical ventilation. It has been repeatedly shown how overdistending alveoli with mechanical ventilation leads to disruption in the alveolar epithelium. Indeed, the extent of pulmonary involvement in patients with ARDS is heterogeneous, and some areas of the lungs are more severely affected than others. This heterogeneity can determine a variable amount of misdistribution of the mechanically delivered tidal volume, so that some alveoli are prone to more distention than others; in fact, it is also generally expected to observe a certain degree of hyperinflation of relatively normally aerated lung regions. Since nonaerated lung tissue is stiffer than normal lung tissue, the total lung compliance is decreased and pressure in the airways is increased. Ventilation with excessive pressures and volumes, with the consequently elevated transpulmonary pressures (i.e., the difference between airway and pleural pressure, which is the pressure effectively distending the lung), play a role in the generation of VILI. In addition, inflation of healthy alveoli close to noninflated, abnormal lung units may generate elevated shear forces that further amplify and contribute to injury of the pulmonary tissue, even if the pressure applied was within safe limits, through a mechanism of “stress amplification”. The effects can be seen at the cellular level, where stretching of the lung beyond its capacity disrupts cellular membranes in the alveoli (7). The consequent cell death then induces inflammation. Moreover, less severe injuries to the cytoskeleton or the extracellular matrix were shown to induce inflammation through a cascade of intracellular signals (8).
The consequences of high-volume ventilation, that is 10 to 15 mL/kg tidal volume, include the development of increased permeability pulmonary edema even in a previously non-injured lung (9) as well as increased formation of edema in the injured lung (10). The first theories developed to explain these negative effects of mechanical ventilation had an overdistention of the alveoli, whereby the injury was mainly attributed to capillary stress failure with a consequent injury to the endothelium and the epithelium. More recent studies showed how ventilation with high tidal volumes may also provoke the initiation of a proinflammatory cascade which then results in lung injury. The structural and functional consequences of this inflammation are practically undistinguishable from those generated by the primary mechanisms underlying lung injury in ARDS. Eventually, high-volume ventilation per se has been shown to lead to a direct release of metalloproteinases (11).
Lung protective ventilation and low tidal volume
Until the late eighties, the conventional approach to mechanical ventilation in ARDS consisted in the use tidal volumes of up to 10–15 mL per kg of body weight (12). Indeed, such volumes were even larger than those in normal, resting subjects (ranging about 7–8 mL/kg, i.e., 500 mL for an average 70-kg patient); however, this was deemed necessary to obtain the normalisation of pCO2 and pH, given the increased CO2 production and the extent of dead space associated with this condition.
In 1993, an American College of Chest Physicians consensus conference recommended a reduction in the applied tidal volume for all patients with ARDS whose plateau pressure was >35 cmH2O, while accepting the consequent possible development of hypercapnia (i.e., the concept of “permissive hypercapnia”) (13). At the time, the level of evidence for such a recommendation was largely based on animal studies, given the lack of human data on the application of a low-tidal-volume strategy and no definitive evidence showing any possible outcome benefit for patients with this approach. However, the physiologic rationale behind the recommendation was strong enough to allow for the translation of that approach into clinical practice.
The first randomized study to test the hypothesis of a potential benefit of protective [i.e., low tidal volume (TV)] ventilation in patients with ARDS was that by Amato et al. in 1998 (14). The authors compared a conventional strategy involving TV of 12 mL/kg, low PEEP and a PCO2 target of 35 to 38 mmHg, with a protective ventilation strategy composed of TV of up to 6 mL/kg, a higher PEEP, and tolerance of hypercapnia. A total of 53 patients were enrolled, and 28-day mortality rate was significantly lower with protective ventilation (38% vs. 71%); a reduced incidence of barotrauma and a significantly higher proportion of patients weaned from ventilation were also seen in the low tidal volume group.
Subsequently, the seminal larger study larger study by the Acute Respiratory Distress Syndrome Network—ARDSNet (the ARMA trial) enrolled 861 patients with ARDS who were randomized to receive mechanical ventilation with either 12 or 6 mL/kg of tidal volume [scaled on predicted body weight (PBW)]; the study showed a significant 22% reduction in mortality in the low tidal volume group (15). Recently, a meta-analysis was conducted by the Cochrane Collaboration including all the RCTs which compared a ventilation strategy based on a lower tidal volume or lower airway pressure (i.e., plateau pressure ≤30 cmH2O) versus a conventional, higher tidal volume strategy. The results showed how 28-day mortality was significantly reduced by the application of a lung-protective strategy (16). In their conclusions, the authors stated how low tidal volume ventilation should become a routine strategy for the treatment of ARDS, deeming it unnecessary to carry out any additional trial. However, despite the publication of the ARMA trial dates back >15 years, the use of a low tidal volume strategy is still not routinely used (17), although the strategy has repeatedly proven to be clinically safe, with no need to increase the dosage of sedatives or neuromuscular blockers to be performed (18,19) .
How to scale tidal volume: PBW vs. airway driving pressure
The rationale behind the use of low TV is that the ventilatable lung parenchyma should not be strained up to an unphysiologic level. Because the strain may be defined as the ratio of TV to the resting lung volume (see below for a more detailed discussion), PBW has been introduced as a surrogate for lung volume since both are related to the patient’s height (20). The formula to obtain PBW is the following: PBW =50.0+0.91 (height in centimeters—152.4) for men; and PBW =45.5+0.91 (height in centimeters—152.4) for women.
The concept underlying this approach is that the actual body weight is not an accurate index of lung size, and since tidal volume should be scaled to the actual lung size, and lung size was shown to be strongly correlated to height and sex (20), PBW was suggested. For example, a person with an ideal weight of 70 kg who then increased his weight by 40 kg still has the same lung size as he/she did when at his ideal weight, and should then be ventilated with a similar tidal volume despite the weight gain.
However, in patients with ARDS, in which inhomogeneity of lung involvement is one of the key features of the clinical picture, the relationship among PBW, resting lung volume, and height is almost lost compared with subjects with healthy lungs (21). Indeed, similar tidal volume can generate far different lung stress/strain in similar subjects (22). Therefore, the same TV/PBW may lead to a completely different strain, depending on the actual amount of ventilatable lung volume still open to ventilation, i.e., the size of the so-called “baby lung” (23). For example, if, in a 70-kg patient with ARDS, the resting lung volume is 300 mL, 6 mL/kg TV/PBW would generate a strain of 420/300 or 1.4; on the other side, if the baby lung volume is 800 mL, the induced strain would be 420/800 or 0.52.
Airway driving pressure has recently been proposed as a scaling factor for a more precise individualisation of the delivered tidal volume (24). Driving pressure has been defined as the ratio of tidal volume by total respiratory system compliance. Its use has been suggested as respiratory system compliance was shown to be related to the total amount of aerated of lung volume (25); as such, its use should be better informative of the extent of lung stress/strain. In a recent pooled analysis conducted on more than 3,500 patients with ARDS managed with varying combinations of tidal volume and PEEP, Amato and coll. found how the parameter which was more strongly associated with an adverse outcome was the airway driving pressure: an increased mortality was only associated with higher plateau pressures in patients with higher driving pressures, whereas the same level of plateau pressure was not associated with increased mortality in case of lower driving pressure. Similarly, elevated PEEP levels exerted a protective effect only if they were associated with low driving pressures; the authors identified a cut-off for increased mortality at a driving pressure of 15 cmH2O (24). A possible conclusion that can be drawn from this study is that TV may be scaled to respiratory system compliance (as a proxy for the amount of ventilatable lung) with the upper limit of safety of the generation of 15 cmH2O of driving pressure. Despite these interesting retrospective findings, the driving pressure approach still has some limitations; in fact the pressure that distends the lung is the transpulmonary pressure, and not the airway pressure. Indeed, this has significant consequences, as it has been shown that chest wall mechanics in patients with ARDS may be unpredictable (26). Conversely, a more appropriate approach from a physiological standpoint could be the assessment of functional residual capacity (FRC), to be used as a proxy of the size of the baby lung. The use of such an approach, so far limited to the research setting, may lead to the design of new studies that may provide evidence for an individualized setting of the tidal volume. As a matter of fact, however, it was recently shown how the use of airway driving pressure in patients with ARDS is an acceptable marker of lung overstretching, as higher values of lung stress, and elastance of the respiratory system and the lung were associated with higher levels of airway driving pressure, whereas patients with lower airway driving pressure had lower levels of stress and partitioned elastance (27).
Lung stress and strain
An injurious strategy of mechanical ventilation leads to development of VILI with a chain of events that starts from a mechanical damage to the lung tissue determined by excess stress-induced strain as the first hit, with a subsequent development of biotrauma as a response to the physical damage caused by the excessive strain (22). In the last years, the group led by Gattinoni applied a bioengineering approach as an analytic tool to assess the effects of mechanical ventilation on the lungs and to determine if progression to lung injury might be delayed or blocked by adjustments in the characteristics of the mechanical breath, with a possible consequent reduction in the incidence of ARDS (28,29).
Stress and strain are useful terms when describing the effect of external forces acting on a subject. When a deformation is applied to the lung skeleton, such as the distending force which drives ventilation into the alveoli (i.e., the transpulmonary pressure), this causes the development of an equal and inverse reacting force, which is termed “stress”. Strain can be defined as the deformation from the resting state in response to an external stress applied to a body, which in the case of the lung are TV and the volume given by PEEP application over the FRC. Thus, global strain is the result of the volume given by tidal ventilation plus that of PEEP. The total strain can then be divided into a dynamic component, i.e., the change in volume change determined by TV over the FRC, and a static component given by the volume change over FRC given by the application of PEEP. Calculations of strain require FRC measurement, while assessment of stress requires the measurement of esophageal pressure as a surrogate for pleural pressure. In fact, the transpulmonary pressure is calculated as the difference between airway and esophageal pressure.
Within physiologic limits, stress and strain are linearly related, so that stress = K × strain, where the proportionality constant K has been termed “specific lung elastance”. Chiumello et al. (22) recently demonstrated that a similar specific lung elastance is present in subjects with healthy lungs as well as in patients with ARDS, averaging 13.5 cmH2O. This means that even during lung injury, barotrauma (stress) and volotrauma (strain) bear the same constant relationship observed in normal subjects.
It was recently found that in pigs, the stress and strain may be lethal in healthy lungs when lung volume reaches total lung capacity (28). The issue, however, is likely more complicated because of the possible regional increase of stress and strain caused by lung inhomogeneity (30), which in turn may act as a “stress riser” that may regionally multiply the applied pressure. However, it seems physiologically sound to suggest that TV is set so that the level of stress (i.e., the end-inspiratory transpulmonary pressure) does not exceed 27 cmH2O, which should correspond to the generation of a strain within the physiologic boundaries of the lung expansion.
Ultra-low tidal volume: the role of extracorporeal support
Patients with ARDS ventilated with a low tidal volume, according to lung-protective ventilation strategy, may still be exposed to forces that can induce/worsen lung injury (31-33); in fact, as mentioned above, applied strain depends on baby-lung size. Several authors postulated that an ultra-protective ventilation strategy based on a further reduction in TV from 6 to 4 mL/kg and plateau pressure from 30 to 25 cmH2O may minimize tidal hyperinflation and attenuate pulmonary inflammation thus improving patient outcome (34,35). However, mechanical ventilation with ultra-low tidal volume and lower plateau pressure may enhance atelectasis onset in some lung regions that may require positive end-expiratory pressure increase to maintain oxygenation (15); moreover, moderate to severe ARDS patients, especially if ventilated with ultra-low tidal volume, may develop hypercapnia with respiratory acidosis. Elevated PCO2, in turn, can lead to increased intracranial pressure, pulmonary hypertension, decreased myocardial contractility, reduced renal blood flow; eventually, this condition may directly bring to release of endogenous catecholamines (36).
Extracorporeal carbon dioxide removal (ECCO2R) may be useful in hypercapnic ARDS patients and may facilitate ultra-low tidal volume ventilation preventing or mitigating respiratory acidosis due to alveolar hypoventilation. During ECCO2R support, metabolic CO2 passing through the extracorporeal membrane lung is removed from patient blood. This decrement is related to fresh gas flow across the membrane while it is relatively independent from blood flow; in fact, with a low blood flow (0.4–1 L/min) obtained with small vascular catheters like the ones used in dialysis, ECCO2R can potentially remove all CO2 produced by metabolism with minimal effect on oxygenation, which instead needs higher blood flows for a significant exchange across the membrane lung to happen (37,38).
Severe ARDS patients may develop life-threatening hypoxemia with or without severe hypercapnia; in this case, extracorporeal membrane oxygenation (ECMO), which use larger catheters and higher blood flow (3–6 L/min), may provide oxygenation as well as carbon dioxide removal and could be used as rescue therapy in these patients (39).
Beyond tidal volume: VILI and mechanical power
VILI arises from repeated application of high mechanical forces that either directly tear a weak tissue or initiate a signaling process that culminates in a pro-inflammatory cascade (6), in the context of an altered lung and in the presence of extrapulmonary factors which may potentially increase the damage. It is generally accepted that VILI arise as a consequence of lung overdistension (volotrauma), alveolar collapse due to insufficient end-expiratory pressure, development of phenomena of pulmonary unit closing and reopening within each breath (atelectrauma), and the consequent development of biotrauma, i.e., the inflammation caused mechanical injuries (40).
Very recently, a new way of looking at the ventilator side of VILI, i.e., the mechanical power, has been introduced (41). According to this approach, every ventilator component already known to be associated to VILI development (tidal volume, driving pressure, RR, inspiratory to expiratory ratio and flow) with the addition of PEEP effect, contribute, each one to his proper extent, to the total amount of energy delivered to the respiratory system (and hence to the lung). The mechanical power concept does not introduce any new component to the field of ventilator-related causes of VILI; instead, it proposes and validates a mathematical description of machine power responsive to the relative contributions of its bedside-adjustable components. Starting from the classical equation of motion, an equation was developed that enables to calculate the mechanical power by some easily obtainable ventilator variables (42). In fact, the external force applied to the pulmonary extracellular matrix times the consequent lung displacement, that corresponds to the product of transpulmonary pressure times the tidal volume, represents the trigger of stress and strain. VILI is thus generated by the cyclic energy load delivered by the ventilator to the respiratory system; this, when applied at a given frequency, may be seen as a sort of “fatigue” of the extracellular matrix (43). If the lung is subject to an “excess” of energy, the unrecovered energy may be enough to lead to a rupture of the molecular bonds of the extracellular matrix polymers (44), to separate from the base membrane cells from the endothelium (45) and the epithelium (46), and to damage the capillary walls (47). The energy load delivered by the ventilator against the respiratory system is composed of a static component, conceptually equivalent to potential energy and consequent to the application of PEEP and the resulting PEEP volume, and a dynamic cyclic component (which correspond to kinetic energy), due to tidal volume above PEEP and the tidal driving pressure. In addition, a resistive and inertial component generated by the pressure spent to move gas, the surface tension and tissue resistances to motion should also be taken into account. Within this framework, energy corresponds to the change in volume times the pressure applied, along the inspiratory pressure-volume relationship.
In a recent paper, Gattinoni et al. (48) demonstrated how a “power equation”, which was derived from the equation of motion of the respiratory system with the addition of PEEP (while inertial forces were neglected), proved to yield comparable values of mechanical power when compared to data obtained experimentally through direct analysis of respiratory tracings. From this equation (49), the pressure (P) in the respiratory system at a given time (t) corresponds to:
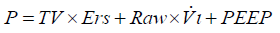
Where Ers is elastance of the respiratory system, Raw is the total resistance of the respiratory system, and Vi the inspiratory flow.
The energy provided by the ventilator per each breath (Ebreath) can be calculated by multiplying in the equation each pressure by the corresponding variation in volume (i.e., TV), as follows:
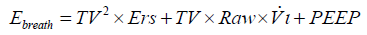
After substituting Vi with TV/Tinsp (the inspiratory time) and then expressing Tinsp as a function of inspiratory-to-expiratory ratio (I:E) and RR, and converting the value to J/min (1 cmH2O ×l = 0.098 J), the following equation may be derived
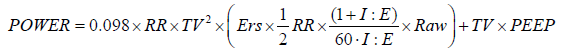
Since it is the interaction between the mechanical power delivered to the portion of pulmonary tissue open to ventilation and the structural characteristics of the latter that lead to development of VILI, it is possible that different combinations of the components of mechanical power, when generating a power greater than a given threshold, may lead to the development of a similar injury. This was recently confirmed by a study in which the lungs of healthy animals were ventilated with different combinations of tidal volume and RR to assess whether a threshold for the development of VILI could be identified (41). The authors found how with values of mechanical power lower than 12 J/min, irrespective of how large was the tidal volume applied, only isolated densities were seen as a consequence of mechanical ventilation at the CT scan. Conversely, when mechanical power was higher than this 12 J/min threshold, all piglets developed whole-lung edema.
Conclusions
In summary, a low tidal volume ventilation strategy proved safe and effective in maintaining gas exchange in critically ill patients without ARDS. In mechanically ventilated patients who are at risk of developing ARDS, high tidal volume ventilation leads to increased incidence of ARDS. In case ARDS is already established, high tidal volumes increase mortality rates. The use of sex and height provides a better estimate of the amount of ventilatable lung when compared to the actual body weight; thus, PBW is still recommended to set the appropriate tidal volume in these patients. The use of airway driving pressure as a scaling factor for tidal volume should overcome the limitations of the use of PBW, namely the heterogeneous nature of ARDS. Newer, physiologically-oriented strategies involving the direct measurement of functional residual capacity as the size of the baby lung may allow to calculate the amount of strain to which the lungs are subject may lead to further optimization of tidal volume in individual patients. Eventually, it has to be kept in mind that tidal volume is not the only factor related to the potential development of VILI, and other ventilator-related parameters should also be considered (namely, plateau pressure, airway driving pressure, RR, inspiratory flow): the promising framework of mechanical power thus seems a potentially more accurate parameter to follow.
Acknowledgements
None.
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- Fan E, Needham DM, Stewart TE. Ventilatory management of acute lung injury and acute respiratory distress syndrome. JAMA 2005;294:2889-96. [Crossref] [PubMed]
- Gattinoni L. Ultra-protective ventilation and hypoxemia. Crit Care 2016;20:130. [Crossref] [PubMed]
- Kallet RH, Hemphill JC 3rd, Dicker RA, et al. The spontaneous breathing pattern and work of breathing of patients with acute respiratory distress syndrome and acute lung injury. Respir Care 2007;52:989-95. [PubMed]
- Nuckton TJ, Alonso JA, Kallet RH, et al. Pulmonary dead-space fraction as a risk factor for death in the acute respiratory distress syndrome. N Engl J Med 2002;346:1281-6. [Crossref] [PubMed]
- Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis 1974;110:556-65. [PubMed]
- Dreyfuss D, Soler P, Basset G, et al. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis 1988;137:1159-64. [Crossref] [PubMed]
- Vlahakis NE, Hubmayr RD. Cellular stress failure in ventilator-injured lungs. Am J Respir Crit Care Med 2005;171:1328-42. [Crossref] [PubMed]
- Ridge KM, Linz L, Flitney FW, et al. Keratin 8 phosphorylation by protein kinase C delta regulates shear stress-mediated disassembly of keratin intermediate filaments in alveolar epithelial cells. J Biol Chem 2005;280:30400-5. [Crossref] [PubMed]
- Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med 1998;157:294-323. [Crossref] [PubMed]
- Bowton DL, Kong DL. High tidal volume ventilation produces increased lung water in oleic acid-injured rabbit lungs. Crit Care Med 1989;17:908-11. [Crossref] [PubMed]
- Pardo A, Selman M, Ridge K, et al. Increased expression of gelatinases and collagenase in rat lungs exposed to 100% oxygen. Am J Respir Crit Care Med 1996;154:1067-75. [Crossref] [PubMed]
- Marini JJ. Evolving concepts in the ventilatory management of acute respiratory distress syndrome. Clin Chest Med 1996;17:555-75. [Crossref] [PubMed]
- Slutsky AS. Mechanical ventilation. American College of Chest Physicians' Consensus Conference. Chest 1993;104:1833-59. [Crossref] [PubMed]
- Amato MB, Barbas CS, Medeiros DM, et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med 1998;338:347-54. [Crossref] [PubMed]
- Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med 2000;342:1301-8. [Crossref] [PubMed]
- Petrucci N, De Feo C. Lung protective ventilation strategy for the acute respiratory distress syndrome. Cochrane Database Syst Rev 2013.CD003844. [PubMed]
- Jaswal DS, Leung JM, Sun J, et al. Tidal volume and plateau pressure use for acute lung injury from 2000 to present: a systematic literature review. Crit Care Med 2014;42:2278-89. [Crossref] [PubMed]
- Cheng IW, Eisner MD, Thompson BT, et al. Acute effects of tidal volume strategy on hemodynamics, fluid balance, and sedation in acute lung injury. Crit Care Med 2005;33:63-70; discussion 239-40. [Crossref] [PubMed]
- Kahn JM, Andersson L, Karir V, et al. Low tidal volume ventilation does not increase sedation use in patients with acute lung injury. Crit Care Med 2005;33:766-71. [Crossref] [PubMed]
- Stocks J, Quanjer PH. Reference values for residual volume, functional residual capacity and total lung capacity. ATS Workshop on Lung Volume Measurements. Official Statement of The European Respiratory Society. Eur Respir J 1995;8:492-506. [Crossref] [PubMed]
- Macnaughton PD, Evans TW. Measurement of lung volume and DLCO in acute respiratory failure. Am J Respir Crit Care Med 1994;150:770-5. [Crossref] [PubMed]
- Chiumello D, Carlesso E, Cadringher P, et al. Lung stress and strain during mechanical ventilation for acute respiratory distress syndrome. Am J Respir Crit Care Med 2008;178:346-55. [Crossref] [PubMed]
- Gattinoni L, Pesenti A. The concept of “baby lung”. Intensive Care Med 2005;31:776-84. [Crossref] [PubMed]
- Amato MB, Meade MO, Slutsky AS, et al. Driving pressure and survival in the acute respiratory distress syndrome. N Engl J Med 2015;372:747-55. [Crossref] [PubMed]
- Gattinoni L, Carlesso E, Cadringher P, et al. Physical and biological triggers of ventilator-induced lung injury and its prevention. Eur Respir J Suppl 2003;47:15s-25s. [Crossref] [PubMed]
- Gattinoni L, Pelosi P, Suter PM, et al. Acute respiratory distress syndrome caused by pulmonary and extrapulmonary disease. Different syndromes? Am J Respir Crit Care Med 1998;158:3-11. [Crossref] [PubMed]
- Chiumello D, Carlesso E, Brioni M, et al. Airway driving pressure and lung stress in ARDS patients. Crit Care 2016;20:276. [Crossref] [PubMed]
- Protti A, Cressoni M, Santini A, et al. Lung stress and strain during mechanical ventilation: any safe threshold? Am J Respir Crit Care Med 2011;183:1354-62. [Crossref] [PubMed]
- Protti A, Andreis DT, Monti M, et al. Lung stress and strain during mechanical ventilation: any difference between statics and dynamics? Crit Care Med 2013;41:1046-55. [Crossref] [PubMed]
- Cressoni M, Chiurazzi C, Gotti M, et al. Lung inhomogeneities and time course of ventilator-induced mechanical injuries. Anesthesiology 2015;123:618-27. [Crossref] [PubMed]
- Terragni PP, Rosboch G, Tealdi A, et al. Tidal hyperinflation during low tidal volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med 2007;175:160-6. [Crossref] [PubMed]
- Bellani G, Guerra L, Musch G, et al. Lung regional metabolic activity and gas volume changes induced by tidal ventilation in patients with acute lung injury. Am J Respir Crit Care Med 2011;183:1193-9. [Crossref] [PubMed]
- Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med 2013;369:2126-36. [Crossref] [PubMed]
- Hager DN, Krishnan JA, Hayden DL, et al. Tidal volume reduction in patients with acute lung injury when plateau pressures are not high. Am J Respir Crit Care Med 2005;172:1241-5. [Crossref] [PubMed]
- Dellinger RP. Positive clinical impact of low tidal volume strategy. Crit Care Med 2005;33:1143-4. [Crossref] [PubMed]
- Feihl F, Perret C. Permissive hypercapnia. How permissive should we be? Am J Respir Crit Care Med 1994;150:1722-37. [Crossref] [PubMed]
- Bein T, Weber-Carstens S, Goldmann A, et al. Lower tidal volume strategy (approximately 3 ml/kg) combined with extracorporeal CO2 removal versus “conventional” protective ventilation (6 ml/kg) in severe ARDS: the prospective randomized Xtravent-study. Intensive Care Med 2013;39:847-56. [Crossref] [PubMed]
- Terragni PP, Del Sorbo L, Mascia L, et al. Tidal volume lower than 6 mL/kg enhances lung protection: role of extracorporeal carbon dioxide removal. Anesthesiology 2009;111:826-35. [Crossref] [PubMed]
- Bein T, Weber F, Philipp A, et al. A new pumpless extracorporeal interventional lung assist in critical hypoxemia/hypercapnia. Crit Care Med 2006;34:1372-7. [Crossref] [PubMed]
- Uhlig U, Uhlig S. Ventilation-induced lung injury. Compr Physiol 2011;1:635-61. [PubMed]
- Cressoni M, Gotti M, Chiurazzi C, et al. Mechanical Power and Development of Ventilator-induced Lung Injury. Anesthesiology 2016;124:1100-8. [Crossref] [PubMed]
- Tonetti T, Cressoni M, Collino F, et al. Volutrauma, Atelectrauma, and Mechanical Power. Crit Care Med 2017;45:e327-e8. [Crossref] [PubMed]
- Protti A, Andreis DT, Milesi M, et al. Lung anatomy, energy load, and ventilator-induced lung injury. Intensive Care Med Exp 2015;3:34. [Crossref] [PubMed]
- Parker JC, Breen EC, West JB. High vascular and airway pressures increase interstitial protein mRNA expression in isolated rat lungs. J Appl Physiol (1985) 1997;83:1697-705. [PubMed]
- Marini JJ, Hotchkiss JR, Broccard AF. Bench-to-bedside review: microvascular and airspace linkage in ventilator-induced lung injury. Crit Care 2003;7:435-44. [Crossref] [PubMed]
- Budinger GR, Sznajder JI. The alveolar-epithelial barrier: a target for potential therapy. Clin Chest Med 2006;27:655-69. abstract ix. [Crossref] [PubMed]
- West JB. Fragility of pulmonary capillaries. J Appl Physiol (1985) 2013;115:1-15. [Crossref] [PubMed]
- Gattinoni L, Tonetti T, Cressoni M, et al. Ventilator-related causes of lung injury: the mechanical power. Intensive Care Med 2016;42:1567-75. [Crossref] [PubMed]
- Marini JJ, Crooke PS 3rd. A general mathematical model for respiratory dynamics relevant to the clinical setting. Am Rev Respir Dis 1993;147:14-24. [Crossref] [PubMed]

