

The molecular epidemiology of hyperphenylalaninemia in Uygur population: incidence from newborn screening and mutational spectra
Introduction
Different from the Han Chinese, the Uygur population originated from the Dingling, Tiele and Uighur ethnic groups, who live in the south of Junggar Basin in Northwest China which is a part of the ancient Silk Road. The Uygur population harbors an extensive genetic admixture of the human population. Newborn screening for hyperphenylalaninemia (HPA) was initiated in 1964 and subsequently adopted in 1981 in mainland China (1). The incidence of HPA is approximately 1/15,000 in newborns worldwide (2). The incidence varies among ethnic and geographical regions. For example, the incidence in Turkey is 1/2,600, Ireland is 1/4,500, China is 1/15,415 (http://zhibao2.xsesc.cn/Login.aspxl), Japan is 1/143,000, and Finland is 1/200,000 (3-6). In Uygur communities with poor economic and medical development, the HPA screening did not begin until 2009. Even today, the screening rate has not yet reached 100%, but a remarkable number of Uygur children have already been diagnosed with PKU in clinics.
HPA is a common autosomal recessive inborn error of amino acid metabolism that primarily results from mutations in the phenylalanine hydroxylase (PAH) gene. About 955 different PAH variants are recorded in database (http://www.biopku.org). The wide variability in the common mutations among ethnic groups and geographical areas makes PAH deficient with great allelic heterogeneity (7). PAH enzyme activity deficiencies result in the inability to convert phenylalanine (Phe) into tyrosine (Tyr), leading to an increased concentration of Phe in the blood and central nervous system. When the blood Phe levels rise above 360 µmol/L, a restrictive low Phe diet needs to be implemented immediately for patients in order to prevent progressively aggravated mental retardation, seizure disorders, and eczema (8). It has been recommended that blood Phe levels need to be controlled between 120 and 600 µmol/L for different age groups (9). So far, a wide array of new treatments such as cell directed therapy, gene therapy and enzyme therapy were performed to PKU patients.
Accordingly, our study aims to systematically analyse the incidence, PAH mutational spectrum, and the follow-up of the Uygur population from January 2010 to December 2016 who live in the southern Junggar Basin in Northwest China. Consequently, we uncovered geographical and ethnic differences in the HPA mutation profile between the places in Northwest China and some other regions and provided guidance for the molecular epidemiology diagnosis of patients with PKU in Uygur population.
Methods
Ethics statement
All procedures performed in this study involving human participants follows the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from individual participants included in the study.
Database and population
We used the National Direct Reporting System for Neonatal Disease Screening database to obtain neonatal birth and screening data from January 2010 to December 2016 (http://zhibao2.xsesc.cn/Login.aspxl). The Uygur screening population data from January 2013 to December 2016 was obtained from the Newborn Disease Screening System of the Xinjiang Uygur Autonomous Region (http://202.85.214.55:5888/login.screen). Also, we manually searched the Uygur screening population registry from January 2010 to December 2012. This study was reviewed and approved by the Human Ethics Committee of the People’s Hospital of Xinjiang Uygur Autonomous Region (2017010).
Sample size calculation
We used the cross-sectional study method to calculate the annual gross incidence rate according to the following formula:
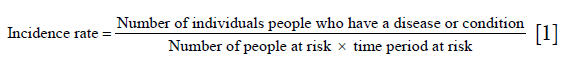
We assume that the word “people” above refers to a group with no relatives included because the number of relatives is negligible as the denominator is very large. Therefore we removed the patients’ relatives from the numerator.

N = number; Z = standard normal distribution boundary value; α=0.05, Zα/2=1.96; α=0.01, Zα/2=2.58; π= expected incidence rate; and δ = admissible error.
Subjects
The Phe levels on dried blood spots were initially quantified using the Guthrie test. The cut off level is 120 µmol/L. When the Phe values were >120 µmol/L at two times, the child would be recalled to our hospital. Tandem mass-spectrometry (TMS), urinary pterin analysis, and determination of DHPR activity were ordered. When the values of Phe at TMS were >120 µmol/L and Phe/Tyr ratio >2.00, it suggested HPA. Then we would determine whether the patients had BH4 deficiency and distinguish the subgroups of them, based on the following indicators, the basic urinary neopterin whose normal value was 0.29–2.61 (mmol/mol Cr), biopterin whose normal value was 0.35–2.67 (mmol/mol Cr), and DHPR whose normal activity was 1.02–3.35 nmol/min/ (5 mmdisc). According to their pretreatment plasma Phe levels, all patients were assigned to one of the three phenotypic categories: Phe levels over 1,200 µmol/L were generally termed “classical Phenylketonuria (cPKU)”, Phe levels of 360–1,200 µmol/L were termed “moderate PKU (mPKU)”, and Phe levels of 120–360 µmol/L were termed “mild hyperphenylalaninemia (MHP)”. Parents and siblings were also investigated to confirm their carrier status.
Treatment and follow-up
Patients with Phe levels above 360 µmol/L under uncontrolled protein intake, should be treated immediately via restricting dietary Phe, while ensuring sufficient calories and protein to meet the needs for children’s growth. According to preliminary screening of Phe concentrations, infants were treated with specialized low Phe milk powder, with low Phe staple food provided after six months and low Phe protein powder could be given from birth to 10 years. We adjusted each child’s diet according to their initial blood Phe concentration and monitored blood Phe for three days after each dietary adjustment. When achieving stable control of Phe levels, monitoring periods would be properly adjusted. A target blood Phe levels of: 120–240 µmol/L was safe for children less than one year old; 120–360 µmol/L was safe for children more than one year old (2,10,11).
Sanger sequencing and capture-based next-generation deep sequencing
Two mL blood samples were collected from patients and their parents. DNA isolation was performed using the QIAamp DNA Blood Mini Kit [250] (QIAGEN, Vienna, Austria). PCR primers were designed to amplify thirteen exons and ten base pairs boundary of the PAH gene. PCR products were sequenced, and data were analyzed using Mutation Survey or Software (SoftGenetics, State College, PA, USA) with the reference to PAH RefSeq NM_000277.1.
Genome DNA was captured using Agilent ClearSeq Inherited Disease kit and sequenced by Illumina XTen system by WuXi NextCODE Genomics Company, and an average coverage of 200× was obtained. Data analyses were performed by the bioinformatics team in our clinical genetic laboratory. Mutations and parental carriers were validated by Sanger sequencing using an ABI 3730 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
Results
Incidence of HPA within the Uygur population
From 2010 to 2016, the screening rate in Uygur population increased from an average of 79% to 83% (Figure 1). A total of 669,832 neonates were screened for HPA, including 580,608 Uygur neonates, 128 non-relative neonates were diagnosed with HPA, including 111 Uygur neonates. In 2010, only 1,969 individuals in Uygur were screened, no HPA patients were detected. From 2011 to 2016, yearly incidence rates varied widely, ranging from 1:1,917 to 1:9,522. The average HPA incidence rate within Uygur population was 1:5,230 (Table 1).


Full table
Demographics of the diagnosed patients
A total of 111 Uygur patients (54 males and 57 females) with HPA were detected. The age of diagnosis ranged from fifty-four days to eight years and the patients were classified based on plasma Phe levels before treatment. Among them, 34.23% (38/111) presented as classical PKU; 50.45% (57/111) had moderate PKU, and 14.41% (16/111) were categorized as MHP and 0.9% (1/111) was presented as BH4 deficiency.
PAH mutational spectrum
PAH variants were detected in 110 individuals among the 111 HPA patients by Sanger sequencing, reaching a 99.1% positive rate. Seven individuals had only one heterozygous PAH mutation been detected (6.37%). Among the seven patients, four have MHP and three have mPKU. Given the recessive inheritance pattern of PAH deficiency and the limited coverage of non-coding regions by current methods, we speculate that another allele mutation might locate in the deep introns or non-coding regions. The one PAH negative case then underwent capture-based next-generation deep sequencing for exploring other possible disease etiologies. Our analysis revealed that this individual has a homozygous mutation in QDPR gene, which is associated with tetrahydrobiopterin deficiency (BH4 deficiency): another manifestation of PKU. The clinical phenotype of BH4 deficiency is more serious than that of PAH deficiency. It’s congruent with the phenotypes of this patient, who showed hypotonia, seizures and mental retardation.
Overall, 58 unique PAH mutations were detected, including thirty-five missense mutations, nine splicing mutations, nine nonsense mutations, one inframe deletion, and one frameshift mutation. Mutations were distributed in all exons except exon 13. Exons 7 had the highest number of mutations (Table 2). A104D in exon 3 was the most prevalent hot spot mutation in the Uygur population (8.41%). Other hot-spot mutations with ten or more than ten alleles in our dataset were marked (Table 2 with &). Comparison of these mutation frequencies based on published literatures (12-20), among countries is shown in Figure 2. R413P and R243Q are the hot spot mutations of Chinese and American population, IVS10-11G > A is the hot spot of Iranian and Turkish. However, R53H and A104D are specific in Uygur patients. Seven known PAH polymorphisms were also detected: IVS2+19T > C (33.3%), IVS4+47C > T (47.8%), IVS4-22C > T (42.4%), IVS9+43G > T (36.1%), p.Q232Q (64%), p.L385L (98.7%), and p.V245V (54.4%).

Full table

Eight novel PAH mutations that had not been previously reported or recorded in the PAHdb or HGMD databases were identified, including two nonsense mutations (c.32T > A, p.L11* and c.590T > A, p.L197*), three missense mutations (c.1102G > A, p.E368K; c.1109A > G, p.E370G; c.1304A > T, p.D435V), one frameshift mutation (c.346-347delGA, p.K115 > Hfs), each of them was detected in one allele; two splicing mutations (c.1200-2A > C, IVS12-2A > C; c.1316-1G > A, IVS13-1G > A), each of them was detected in seven alleles (Table 2 with #), distributing in twenty patients (three patients were homozygous of IVS13-1G > A mutations, others were compound heterozygous mutations). Three missense mutations were all predicted to be pathogenic using SIFT, PolyPhen 2, and MutationTaster software. All of the IVS12-2A > C mutation was found in classical PKU and combination with IVS10-11G > A, A104D, P147L, which was mostly found in classical PKU. Two nonsense mutations (L11* and L197*) and missense mutations (E368K) were found in moderate PKU. Two missense mutations (E370G and D435V) were found in MHP.
Correlation between genotype and phenotype in Uygur HPA patients
A total of 110 Uygur patients carrying PAH mutations and one QDPR mutations constituted a spectrum of 58 different genotypic combinations (Table S1). The genotypes of PAH were divided into homozygous (n=36) and heterozygous (n=73). Variant A104D appeared in the most forms of homozygous. A total of 14 patients carried three variants, 7 of them were combination of 6 homozygous of A104, R413P, L430PC, IVS10-11G > A, IVS4+5G > T, I421T. A total of six patients carried only one variant, four of whom were in MHP and two were in mPKU.

Full table
Identification of the PAH gene mutation in patients from two families in a village
A Uygur village in Akesu has a population of about 2,000 with approximately 60 births every year. Villagers do not intermarry with other villages or other ethnic groups. The R413P mutation was identified in four patients from two families (Figure 3). The two families had no relationship but the family members were cousins, and all the patients had symptoms of PKU. As previously described, this mutation is a hot spot mutation within the Uygur population. Sixteen of 34 members of these two families carried R413P heterozygous mutations with an alarming carrier rate of approximately 1:2.1.

Discussion
Over the last 60 years, researches on the pathophysiology and treatment of PKU have progressed rapidly all over the world. However, similar analyses and investigation within the Uygur population have been lagging until now. Our data provide the first accurate assessment of PKU incidence within the Uygur population and highlight the genotypes and frequency of PAH mutations in this community.
The screening rates of HPA incidence fluctuated from 2011 to 2014. Such fluctuations could be affected by multiple factors, leading to variations in the incidence rates (21). In 2014, however, an unexpectedly low rate of HPA incidence was logged, likely due to a change in the screening system, from paper-based to electronic. We speculate that the reform was not so thoroughly executed at the beginning and consequently, some MHP patients were not recalled. HPA incidence rates within the Uygur population were relatively stable in 2015 and 2016, with an estimated rate of 1:5,230, which is consistent with the average rate across the entire period from 2010 to 2016. This rate is higher than that all around China (1:15,415).
Eight novel variants were identified in this study. Two variants were identified as splice variant IVS12-2A > C located in intron 12 and IVS13-1G > A located in intron 13. The bases at the junctions of the introns and exons are AG/GT. The mutation is located at the boundaries of the intron/exon and may cause abnormal RNA splicing. The remaining seven mutations were predicted to be pathogenic using mutation analysis software. The allele counts of top mutations are ten or more than ten (Table 2 with &). Herein, the hot spot mutation of the R413Pand R243Q is also common in Chinese, Japanese, and American population, and IVS10-11G > A is also a common hot spot mutation among Iranian and Turkish. However, R53H and A104D are specific in Uygur patients.
The Uygur population originated from the Dingling, Tiele and Uighur ethnic groups and were socialized by Turk in 840–1212 Anno Domini (AD) with distinct culture characteristics driven by the ancient Silk Road, an important gallery that connected culture and trade among China, Asia, and Europe. At present, approximately 11.3 million of Uygur live in oasis of Tarim Basin, in the south of Tianshan mountains in Xinjiang (22). They have their own languages, eating habits, and national costumes. Uygur population community is on the region of Silk Road, and the significant genetic admixing may occur as a consequence of ancestral migration to this region.
The exclusivity marriage in this population makes a high incidence of some hereditary diseases. In a village with 2,000 people, four patients from two families were found symptoms of PKU. The mutation site is the hot spot of R413P. The carrier rate is approximately 1:2.1. Uygur have a relatively lower rate of DHPR deficiencies, compared with that in whole China, Turkey and Iran. Only one patient of QDPR mutation was detected. However, this study may miss another form of BH4 deficiency, GTP cyclohydrolase (GTPCH-deficient) by newborn screening when the blood Phe levels were not increased (23).
This is the first study to investigate the HPA incidence rate within a large Uygur population and the incidence rate is significantly high. The data also highlight the regional differences in PAH genotypes, suggesting not only a consanguineous relationship, but also distinct differences between Asian and Caucasian populations.
Acknowledgments
We are very grateful to the patients and their families as well as the clinicians taking care of the patients, and our genetics laboratory teams who contributed to this study.
Funding: This study was funded by the National Natural Science Foundation of China (81741102), the Natural Science Foundation of Xinjiang Province (2016 D01C116), and the Shanghai Key Laboratory of Birth Defects (13DZ2260600).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
Ethical Statement: The study was approved by the Human Ethics Committee of the People’s Hospital of Xinjiang Uygur Autonomous Region (2017010) and written informed consent was obtained from all patients.
References
- Shi XT, Cai J, Wang YY, et al. Newborn screening for inborn errors of metabolism in mainland china: 30 years of experience. JIMD Rep 2012;6:79-83. [Crossref] [PubMed]
- Mitchell JJ, Trakadis YJ, Scriver CR. Phenylalanine hydroxylase deficiency. Genet Med 2011;13:697-707. [Crossref] [PubMed]
- Aoki K, Wada Y. Outcome of the patients detected by newborn screening in Japan. Acta Paediatr Jpn 1988;30:429-34. [Crossref] [PubMed]
- Guldberg P, Henriksen KF, Sipila I, et al. Phenylketonuria in a low incidence population: molecular characterisation of mutations in Finland. J Med Genet 1995;32:976-8. [Crossref] [PubMed]
- Tezel B, Dilli D, Bolat H, et al. The development and organization of newborn screening programs in Turkey. J Clin Lab Anal 2014;28:63-9. [Crossref] [PubMed]
- Zschocke J, Mallory JP, Eiken HG, et al. Phenylketonuria and the peoples of Northern Ireland. Hum Genet 1997;100:189-94. [Crossref] [PubMed]
- Murad H, Dabboul A, Moassas F, et al. Mutation spectrum of phenylketonuria in Syrian population: genotype-phenotype correlation. Gene 2013;528:241-7. [Crossref] [PubMed]
- Flydal MI, Martinez A. Phenylalanine hydroxylase: function, structure, and regulation. IUBMB Life 2013;65:341-9. [Crossref] [PubMed]
- Yano S, Moseley K, Fu X, et al. Evaluation of Tetrahydrobiopterin Therapy with Large Neutral Amino Acid Supplementation in Phenylketonuria: Effects on Potential Peripheral Biomarkers, Melatonin and Dopamine, for Brain Monoamine Neurotransmitters. PLoS One 2016;11:e0160892. [Crossref] [PubMed]
- Blau N. Genetics of Phenylketonuria: Then and Now. Hum Mutat 2016;37:508-15. [Crossref] [PubMed]
- Singh RH, Rohr F, Frazier D, et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet Med 2014;16:121-31. [Crossref] [PubMed]
- Aulehla-Scholz C, Heilbronner H. Mutational spectrum in German patients with phenylalanine hydroxylase deficiency. Hum Mutat 2003;21:399-400. [Crossref] [PubMed]
- Dobrowolski SF, Heintz C, Miller T, et al. Molecular genetics and impact of residual in vitro phenylalanine hydroxylase activity on tetrahydrobiopterin responsiveness in Turkish PKU population. Mol Genet Metab 2011;102:116-21. [Crossref] [PubMed]
- Kostandyan N, Britschgi C, Matevosyan A, et al. The spectrum of phenylketonuria genotypes in the Armenian population: identification of three novel mutant PAH alleles. Mol Genet Metab 2011;104 Suppl:S93-6. [Crossref] [PubMed]
- Lee DH, Koo SK, Lee KS, et al. The molecular basis of phenylketonuria in Koreans. J Hum Genet 2004;49:617-21. [Crossref] [PubMed]
- Okano Y, Kudo S, Nishi Y, et al. Molecular characterization of phenylketonuria and tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency in Japan. J Hum Genet 2011;56:306-12. [Crossref] [PubMed]
- Ramus SJ, Treacy EP, Cotton RG. Characterization of phenylalanine hydroxylase alleles in untreated phenylketonuria patients from Victoria, Australia: origin of alleles and haplotypes. Am J Hum Genet 1995;56:1034-41. [PubMed]
- Santos LL, Castro-Magalhaes M, Fonseca CG, et al. PKU in Minas Gerais State, Brazil: mutation analysis. Ann Hum Genet 2008;72:774-9. [Crossref] [PubMed]
- Zare-Karizi S, Hosseini-Mazinani SM, Khazaei-Koohpar Z, et al. Mutation spectrum of phenylketonuria in Iranian population. Mol Genet Metab 2011;102:29-32. [Crossref] [PubMed]
- Zhu T, Ye J, Han L, et al. Variations in genotype-phenotype correlations in phenylalanine hydroxylase deficiency in Chinese Han population. Gene 2013;529:80-7. [Crossref] [PubMed]
- Tu WJ, Cai J, Shi XD. Newborn screening for inborn errors of metabolism in Beijing, China: 22 years of experience. J Med Screen 2011;18:213-4. [Crossref] [PubMed]
- Years and Ethnic population. Statistic Bureau of Xinjiang Uygur Autonomous Region.
- Blau N, Martinez A, Hoffmann GF, et al. DNAJC12 deficiency: A new strategy in the diagnosis of hyperphenylalaninemias. Mol Genet Metab 2018;123:1-5. [Crossref] [PubMed]

